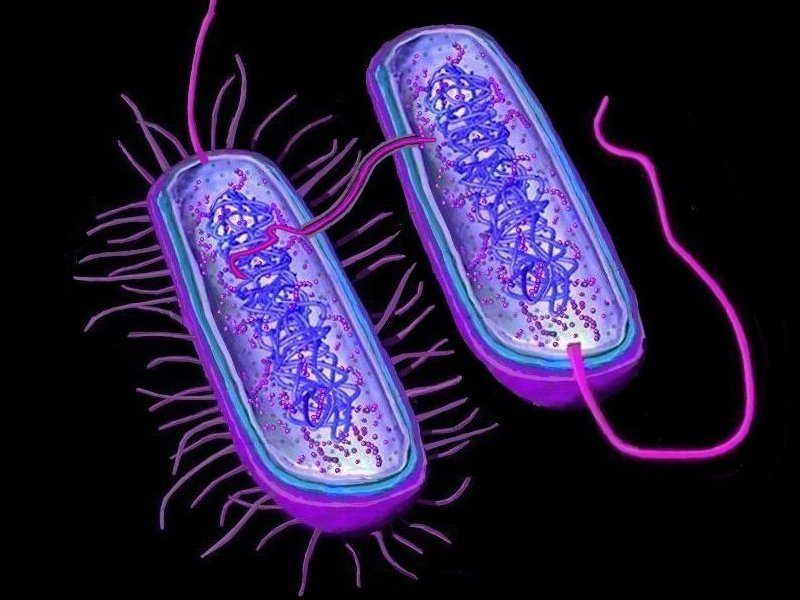
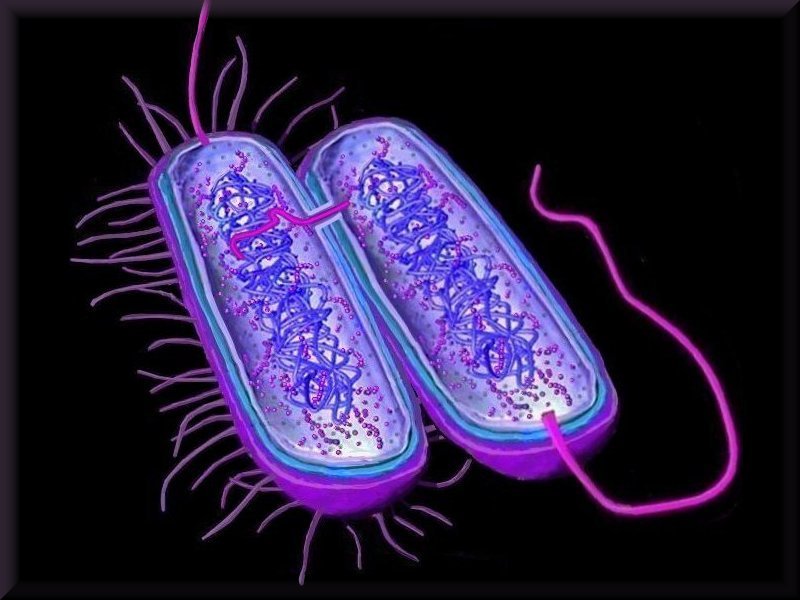
 Horizontal gene transfer
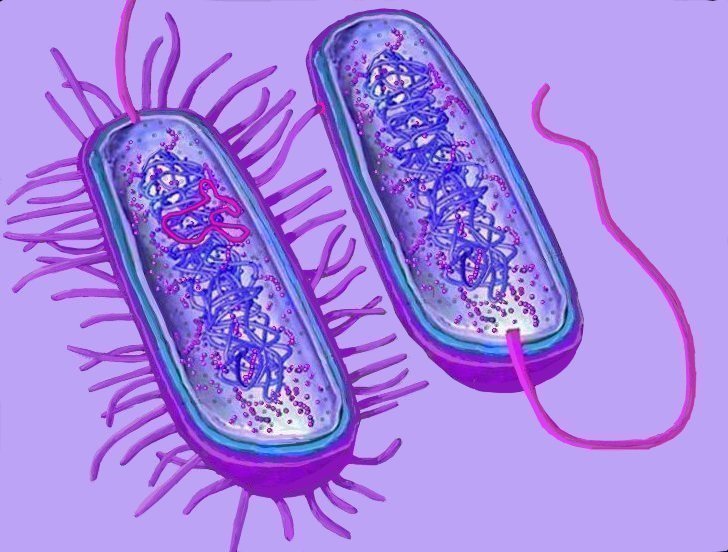
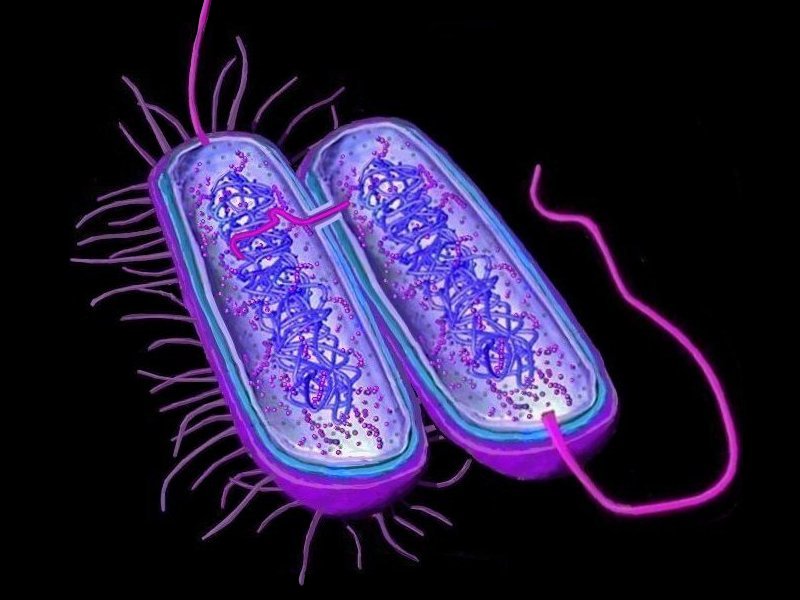
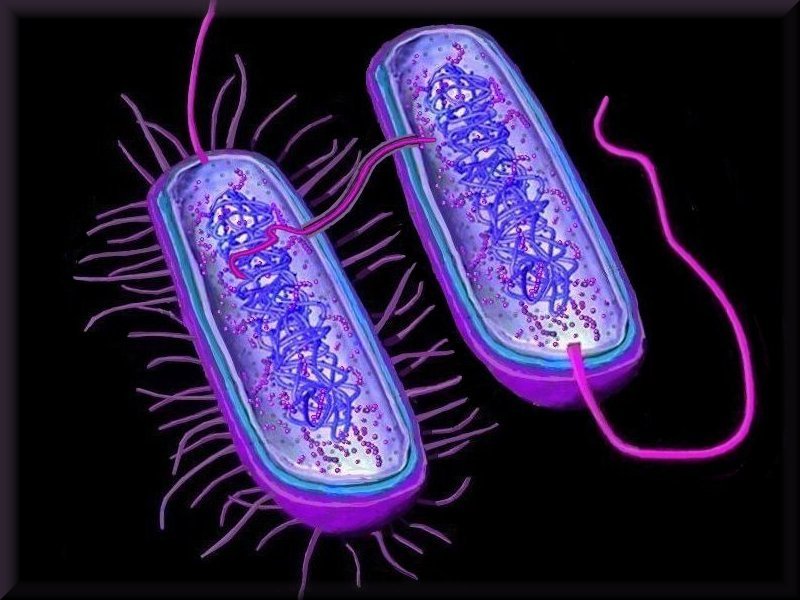
Horizontal gene transfer - gene swapping - has blurred the evolutionary relationships (
phylogeny) of prokaryotes (left,
adapted - click to enlarge), and continues to provide a mechanism for the sharing of antibiotic resistance between bacteria. (see
The net of life: Reconstructing the microbial phylogenetic networkV. Kunin, L. Goldovsky, N. Darzentas, and C. A. OuzounisGenome Res. 1 July 2005.
pdf)
Three mechanisms of horizontal (lateral) gene transfer are recognized: direct bacterial
conjugation, bacteriophage mediated
transduction between bacteria, and bacterial
transformation by uptake of DNA fragments.
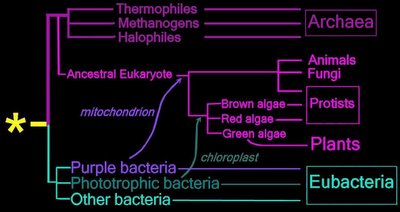
A major form of
vertical gene transfer followed
serial endosymbiotic events, in which ingested purple bacteria and Cyanobacteria became eukaryotic
mitochondria and
chloroplasts respectively. The ingested prokaryotes are believed to have relinquished certain genes to the nuclei of their host cells, a process known as
endosymbiotic gene transfer.
Horizontal Genomics: Mobile genetic elements: the agents of open source evolution. Mobile genetic elements: the agents of open source evolution.: "Horizontal genomics is a new field in prokaryotic biology that is focused on the analysis of DNA sequences in prokaryotic chromosomes that seem to have originated from other prokaryotes or eukaryotes. However, it is equally important to understand the agents that effect DNA movement: plasmids, bacteriophages and transposons. Although these agents occur in all prokaryotes, comprehensive genomics of the prokaryotic mobile gene pool or 'mobilome' lags behind other genomics initiatives owing to challenges that are distinct from cellular chromosomal analysis. Recent work shows promise of improved mobile genetic element (MGE) genomics and consequent opportunities to take advantage - and avoid the dangers - of these 'natural genetic engineers'. This review describes MGEs, their properties that are important in horizontal gene transfer, and current opportunities to advance MGE genomics."
Frost LS,
Leplae R,
Summers AO,
Toussaint A. Mobile genetic elements: the agents of open source evolution.
Nat Rev Microbiol. 2005 Sep;3(9):722-32.Nat Rev Microbiol. 2005 Sep;3(9):722-32.
The Balance of Driving Forces During Genome Evolution in Prokaryotes.
Genomes are shaped by evolutionary processes such as gene genesis, horizontal gene transfer (HGT), and gene loss. To quantify the relative contributions of these processes, we analyze the distribution of 12,762 protein families on a phylogenetic tree, derived from entire genomes of 41 Bacteria and 10 Archaea. We show that gene loss is the most important factor in shaping genome content, being up to three times more frequent than HGT, followed by gene genesis, which may contribute up to twice as many genes as HGT. We suggest that gene gain and gene loss in prokaryotes are balanced; thus, on average, prokaryotic genome size is kept constant. Despite the importance of HGT, our results indicate that the majority of protein families have only been transmitted by vertical inheritance. To test our method, we present a study of strain-specific genes of Helicobacter pylori, and demonstrate correct predictions of gene loss and HGT for at least 81% of validated cases. This approach indicates that it is possible to trace genome content history and quantify the factors that shape contemporary prokaryotic genomes.
Victor Kunin and Christos A. Ouzounis The Balance of Driving Forces During Genome Evolution in Prokaryotes Genome Research 13:1589-1594, 2003
Full Text Full Text (PDF)Examining bacterial species under the specter of gene transfer and exchange.
Even in lieu of a dependable species concept for asexual organisms, the classification of bacteria into discrete taxonomic units is considered to be obstructed by the potential for lateral gene transfer (LGT) among lineages at virtually all phylogenetic levels. In most bacterial genomes, large proportions of genes are introduced by LGT, as indicated by their compositional features and/or phylogenetic distributions, and there is also clear evidence of LGT between very distantly related organisms. By adopting a whole-genome approach, which examined the history of every gene in numerous bacterial genomes, we show that LGT does not hamper phylogenetic reconstruction at many of the shallower taxonomic levels. Despite the high levels of gene acquisition, the only taxonomic group for which appreciable amounts of homologous recombination were detected was within bacterial species. Taken as a whole, the results derived from the analysis of complete gene inventories support several of the current means to recognize and define bacterial species.
Howard Ochman, Emmanuelle Lerat, and Vincent Daubin Examining bacterial species under the specter of gene transfer and exchange PNAS May 3, 2005 vol. 102 Suppl. 1 6595-6599
Full Text Full Text (PDF)Adaptive evolution of bacterial metabolic networks by horizontal gene transfer.: "Numerous studies have considered the emergence of metabolic pathways, but the modes of recent evolution of metabolic networks are poorly understood. Here, we integrate comparative genomics with flux balance analysis to examine (i) the contribution of different genetic mechanisms to network growth in bacteria, (ii) the selective forces driving network evolution and (iii) the integration of new nodes into the network. Most changes to the metabolic network of Escherichia coli in the past 100 million years are due to horizontal gene transfer, with little contribution from gene duplicates. Networks grow by acquiring genes involved in the transport and catalysis of external nutrients, driven by adaptations to changing environments. Accordingly, horizontally transferred genes are integrated at the periphery of the network, whereas central parts remain evolutionarily stable. Genes encoding physiologically coupled reactions are often transferred together, frequently in operons. Thus, bacterial metabolic networks evolve by direct uptake of peripheral reactions in response to changed environments."
Pal C,
Papp B,
Lercher MJ. Adaptive evolution of bacterial metabolic networks by horizontal gene transfer.
Nat Genet. 2005 Dec;37(12):1372-5. Epub 2005 Nov 20.
Horizontal gene transfer in bacterial and archaeal complete genomes.
There is growing evidence that horizontal gene transfer is a potent evolutionary force in prokaryotes, although exactly how potent is not known. We have developed a statistical procedure for predicting whether genes of a complete genome have been acquired by horizontal gene transfer. It is based on the analysis of G+C contents, codon usage, amino acid usage, and gene position. When we applied this procedure to 17 bacterial complete genomes and seven archaeal ones, we found that the percentage of horizontally transferred genes varied from 1.5% to 14.5%. Archaea and nonpathogenic bacteria had the highest percentages and pathogenic bacteria, except for Mycoplasma genitalium, had the lowest. As reported in the literature, we found that informational genes were less likely to be transferred than operational genes. Most of the horizontally transferred genes were only present in one or two lineages. Some of these transferred genes include genes that form part of prophages, pathogenecity islands, transposases, integrases, recombinases, genes present only in one of the two Helicobacter pylori strains, and regions of genes functionally related. All of these findings support the important role of horizontal gene transfer in the molecular evolution of microorganisms and speciation.
Garcia-Vallve S,
Romeu A,
Palau J. Horizontal gene transfer in bacterial and archaeal complete genomes.
Genome Res. 2000 Nov;10(11):1719-25.
Free Full Text Article.
Stochastic Models for Horizontal Gene Transfer : Taking a Random Walk Through Tree Space.
Horizontal gene transfer (HGT) plays a critical role in evolution across all domains of life with important biological and medical implications. I propose a simple class of stochastic models to examine HGT using multiple orthologous gene alignments. The models function in a hierarchical phylogenetic framework. The top level of the hierarchy is based on a random walk process in "tree space" that allows for the development of a joint probabilistic distribution over multiple gene trees and an unknown, but estimable species tree. I consider two general forms of random walks. The first form is derived from the subtree prune and regraft (SPR) operator that mirrors the observed effects that HGT has on inferred trees. The second form is based on walks over complete graphs and offers numerically tractable solutions for an increasing number of taxa. The bottom level of the hierarchy utilizes standard phylogenetic models to reconstruct gene trees given multiple gene alignments conditional on the random walk process. I develop a well-mixing Markov chain Monte Carlo algorithm to fit the models in a Bayesian framework. I demonstrate the flexibility of these stochastic models to test competing ideas about HGT by examining the complexity hypothesis. Using 144 orthologous gene alignments from six prokaryotes previously collected and analyzed, Bayesian model selection finds support for (1) the SPR model over the alternative form, (2) the 16S rRNA reconstruction as the most likely species tree, and (3) increased HGT of operational genes compared to informational genes.
Marc A. Suchard Stochastic Models for Horizontal Gene Transfer Taking a Random Walk Through Tree Space Genetics, Vol. 170, 419-431, May 2005
Full Text Full Text (PDF)A sensitive, support-vector-machine method for the detection of horizontal gene transfers in viral, archaeal and bacterial genomes.
In earlier work, we introduced and discussed a generalized computational framework for identifying horizontal transfers. This framework relied on a gene's nucleotide composition, obviated the need for knowledge of codon boundaries and database searches, and was shown to perform very well across a wide range of archaeal and bacterial genomes when compared with previously published approaches, such as Codon Adaptation Index and C + G content. Nonetheless, two considerations remained outstanding: we wanted to further increase the sensitivity of detecting horizontal transfers and also to be able to apply the method to increasingly smaller genomes. In the discussion that follows, we present such a method, Wn-SVM, and show that it exhibits a very significant improvement in sensitivity compared with earlier approaches. Wn-SVM uses a one-class support-vector machine and can learn using rather small training sets. This property makes Wn-SVM particularly suitable for studying small-size genomes, similar to those of viruses, as well as the typically larger archaeal and bacterial genomes. We show experimentally that the new method results in a superior performance across a wide range of organisms and that it improves even upon our own earlier method by an average of 10% across all examined genomes. As a small-genome case study, we analyze the genome of the human cytomegalovirus and demonstrate that Wn-SVM correctly identifies regions that are known to be conserved and prototypical of all beta-herpesvirinae, regions that are known to have been acquired horizontally from the human host and, finally, regions that had not up to now been suspected to be horizontally transferred. Atypical region predictions for many eukaryotic viruses, including the -, ß- and -herpesvirinae, and 123 archaeal and bacterial genomes, have been made available online at
http://cbcsrv.watson.ibm.com/HGT_SVM/.
Aristotelis Tsirigos and Isidore Rigoutsos, A sensitive, support-vector-machine method for the detection of horizontal gene transfers in viral, archaeal and bacterial genomes. Nucleic Acids Research 2005 33(12):3699-3707; doi:10.1093/nar/gki660
Full Text Free
The fate of laterally transferred genes: Life in the fast lane to adaptation or death.
Large-scale genome arrangement plays an important role in bacterial genome evolution. A substantial number of genes can be inserted into, deleted from, or rearranged within genomes during evolution. Detecting or inferring gene insertions/deletions is of interest because such information provides insights into bacterial genome evolution and speciation. However, efficient inference of genome events is difficult because genome comparisons alone do not generally supply enough information to distinguish insertions, deletions, and other rearrangements. In this study, homologous genes from the complete genomes of 13 closely related bacteria were examined. The presence or absence of genes from each genome was cataloged, and a maximum likelihood method was used to infer insertion/deletion rates according to the phylogenetic history of the taxa. It was found that whole gene insertions/deletions in genomes occur at rates comparable to or greater than the rate of nucleotide substitution and that higher insertion/deletion rates are often inferred to be present at the tips of the phylogeny with lower rates on more ancient interior branches. Recently transferred genes are under faster and relaxed evolution compared with more ancient genes. Together, this implies that many of the lineage-specific insertions are lost quickly during evolution and that perhaps a few of the genes inserted by lateral transfer are niche specific.
Weilong Hao and G. Brian Golding The fate of laterally transferred genes: Life in the fast lane to adaptation or death Genome Research 16:636-643, 2006
GENSTYLE: exploration and analysis of DNA sequences with genomic signature
Full Text free











































{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}